


- Presentación
- Clasificación cardiopatías congénitas
- Epidemiología y patogenia
- Soplos orgánicos y soplo funcional
- Repercusión clínica de las cardiopatías congénitas
- Tratamiento de las cardiopatías congénitas
- Cardiopatías acianógenas con cortocircuito izquierda-derecha
- Cardiopatías acianógenas sin cortocircuito
- Definición cianosis. Diferencia hipoxia – cianosis
- Clasificación cardiopatías cianosantes
- Tetralogia de Fallot
- Atresia pulmonar con Comunicación interventricular
- D-Transposición de grandes arterias
- Corazón univentricular
- Hipertensión pulmonar. Síndrome de Eisenmenger
- Enfermedades del pericardio, miocardio y endocardio
- Enfermedad de Kawasaki
- ECG en el niño
- Arritmias en pediatría
El avance en las técnicas cardiacas diagnósticas, quirúrgicas y en el manejo médico del recién nacido con patología grave, han permitido una mejora en la supervivencia del recién nacido y del niño con cardiopatía congénita. En la actualidad el 85% llega a edad adulta.
Todas las cardiopatías congénitas tienen en la actualidad una posibilidad de actuación terapéutica y en casos aislados de fallo ventricular primario el trasplante cardiaco es una opción factible también en edad pediátrica.
La cardiopatía congénita es una alteración en la estructura del corazón producida durante el desarrollo embriológico cardiaco, que afecta a su funcionamiento. Además, el niño puede presentar una serie de enfermedades cardiovasculares, algunas adquiridas que requieren conocimientos fisiopatológicos para su reconocimiento.
Este apartado de nivel básico va dirigido a estudiantes de Medicina, profesionales del ámbito sanitario relacionados con la pediatría y con pacientes con cardiopatías congénitas, pediatras y cardiólogos en formación quienes quieran entender las cardiopatías congénitas.
Lecturas recomendadas
Existen múltiples y variadas anomalías cardiacas, con posibilidad de combinación de unas con otras, lo que hace que su número se incremente considerablemente. No hay dos cardiopatías congénitas (CC) idénticas, pero sí modelos similares.
Varios defectos estructurales pueden estar asociados e incluyen una única cardiopatía que corresponde a un mismo fallo embriológico que explica todas las malformaciones (como la Tetralogía de Fallot), aunque otras son más complejas y afectan a varias vías embriológicas ocasionando afectación en diferentes segmentos y cavidades cardiacas
En la actualidad las CC se clasifican según:
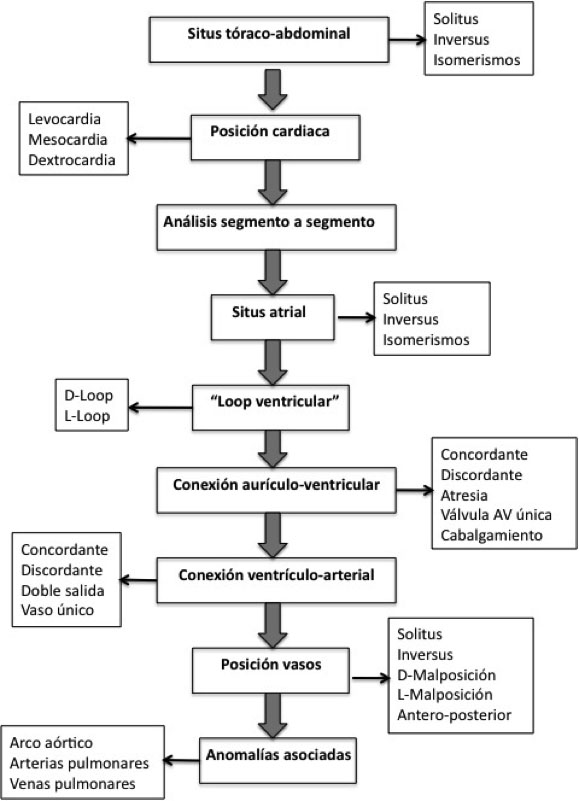
- Anatomía: (más propia de los cardiólogos): la más utilizada hoy en día es la del análisis segmentario, de Robert Anderson.
Las CC se describen definiendo la morfología de las aurículas, ventrículos, válvulas aurículo-ventriculares y posición de los vasos.

Figura 1: Análisis segmentario del corazón<
El corazón normal está en posición denominada Situs solitus, en levocardia y levoápex (corazón situado en hemitórax izquierdo y con la punta a la izquierda), con concordancia aurículo-ventricular y ventrículo-arterial (la aurícula derecha conecta con el ventrículo derecho y éste con la arteria pulmonar).
La disposición de los vasos o arterias puede ser normal (aorta posterior y pulmonar anterior); en d-transposición (aorta anterior y a la derecha); l-transposición (aorta anterior y a la izquierda) o vasos en posición lado a lado.
Existe otra clasificación basada en el desarrollo embriológico del tubo cardiaco, es la descrita por el Dr. Van Praagh (escuela americana). En la clasificación de Van Praagh es importante la embriología cardiaca y es característica la representación de la anatomía cardiaca con 3 letras: El corazón normal se representa como ⎨S,D,S⎬
- 1ª Letra: corresponde al situs visceroatrial: “S” situs solitus, “I” situs inversus y “A” en los Isomerismos (heteroataxias, antes llamados Situs ambiguos).
- 2ª Letra: dirección del giro del tubo cardiaco, “D” si es normal (D-loop, a la derecha); “L”; si está invertido (L-loop, izquierda) y “X” si es indeterminado.
- 3ª Letra: situación de los vasos, “S” en posición normal con pulmonar anterior a la aorta, “I” la aorta colocada a la izquierda por la pulmonar en el situs inversus, “D” si la aorta esta anterior y derecha, y “L” si es anterior e izquierda.
- Fisiopatologia (o funcional): se clasifican según su presentación clínica más característica. Se dividen en acianógenas, cianosantes o formas mixtas (pueden presentar cianosis según la cantidad de mezcla y aumento del flujo pulmonar).

Robert Anderson: Anderson R.H; Ann Thorac Surg 1996: 62: 710-716
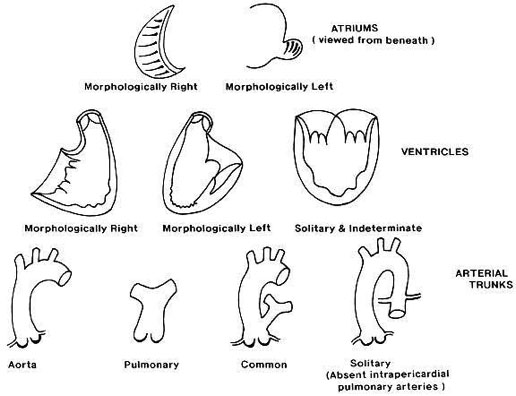
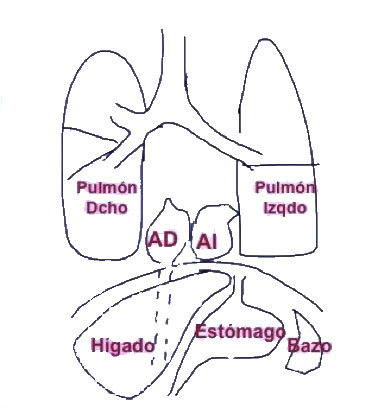
Situs solitus
El situs visceral es el patrón de asimetría del cuerpo, en el hemicuerpo derecho está el hígado y la vena cava inferior (VCI), el apéndice, el intestino delgado y el bronquio derecho que es más corto, dado que se bifurca antes que el izquierdo. En el lado izquierdo están el bazo, el estómago y el colon. El corazón está dentro del tórax situado a la izquierda y con la punta dirigida a la izquierda (levocardia y levoápex). Las aurículas se definen por sus orejuelas, la aurícula derecha (AD) es triangular y la izquierda es en dedo de guante, las orejuelas definen el situs, en general a la AD llega la VCI (ojo, a veces hay agenesia de VCI), y a la aurícula izquierda llegan las venas pulmonares. Cuando la triada hígado-VCI-AD está a la derecha hablamos de situs solitus
Las CC son en la actualidad las malformaciones más frecuentes causantes de la mayor mortalidad infantil antes del año de vida. En general, afectan a entre el 5 al 10 por mil de los RN vivos, de los cuales cerca de la mitad tiene una cardiopatía clínicamente significativa y un tercio de ellos pueden precisar cirugía cardiaca correctora en edades precoces, sobre todo en período neonatal.
Los estudios epidemiológicos sugieren que existen factores genéticos que contribuyen al desarrollo de las CC
En casos con antecedentes familiares de CC los riesgos de recurrencia son variables dependiendo del tipo de cardiopatía, si corresponde a malformación aislada o asociada a un síndrome, si existe enfermedad materna asociada (diabetes) o exposición a teratógenos (rubeola, alcohol, drogas, etc).
El sistema cardiovascular en el ser humano comienza a aparecer hacia la mitad de la tercera semana del desarrollo intrauterino, cuando el embrión ya no es capaz de satisfacer sus necesidades nutricionales por mecanismos primitivos.
Cualquier proceso que afecte a las fases de la cardiogénesis puede originar anomalías estructurales cardiacas. Muchas de las cardiopatías tienen un origen embriológico común, de ahí que una misma cardiopatía incluya varios defectos cardiacos.
Si quieres saber más sobre la embriología cardiaca, lee este articulo Pediatr Cardiol 2004;25:191-200, y mira las siguientes presentaciones on-line
Existen factores genéticos que contribuyen al desarrollo de las cardiopatías, entre ellos alteraciones cromosómicas numéricas (trisomías, monosomías), anomalías cromosómicas estructurales (microdeleciones), mutaciones de uno o un par de genes (autosómica dominante, recesiva o ligada al cromosoma X) y mutaciones aisladas por factores ambientales en interacción (herencia multifactorial) (Ver Tabla 1). Pueden encontrarse acompañando a malformaciones de otros órganos (más frecuente en defectos genéticos) o aisladas (más asociadas a un patrón de herencia multifactorial).
El estudio de las enfermedades congénitas en la actualidad está basado en la detección de mutaciones o polimorfismos de genes de los sujetos afectos.
| Riego % | |
|---|---|
| Población general | 0,5-1% |
| Familiar 1ª grado afectado | 2-3% |
| Dos hermanos o hijo y padre/madre | 10% |
| Más de 2 parientes de 1er grado | Alrededor 50% |
Tabla 1 Riesgo de Cardiopatías congénitas en población normal, en familiares con casos afectos y Síndromes genéticos más frecuentes asociados a cardiopatías
| SINDROME % | CARDIOPATÍA MÁS FRECUENTE ASOCIADA | ||
|---|---|---|---|
| CROMOSOMOPATÍAS | Down (trisomía 21) | 50% | CAV, CIV, CIA, PCA |
| Edward (trisomía 18) | 100% | CIV, CIA, PCA | |
| Patau (trisomía 13) | 100% | CIV, CIA, PCA | |
| Turner (monosomía X) | 25% | CoA, AoB, EA, MHC | |
| MICRODELECCIONES | Delección 9p | Frecuente | CIV, PCA, EP |
| Delección 4p | Frecuente | CIA, EP, CIV | |
| Delección 22q11 (SDM DiGeorge) | 90% | CIV, AAD, TF, EP, TAC | |
| Delección 7q11 (SDM Williams-Beuren) | 100% | EA supravalvular, ETP | |
| MUTACIONES | Noonan (PTPN11) | 90% | EP, MHC, CIA, CIV |
| Holt-Oram (TXB5) | 90% | CIA, BAV CIA, CAV | |
| Alagille (JAG1) | 95% | ERAP, TF, CIA, CIV | |
| SDM de Marfan (FBN1) | Frecuente | IAo, Aneurisma Ao, ProM |
Abreviaturas:
- AAD arco aórtico derecho
- Ao aorta
- AoB aorta bicúspide
- BAV bloqueo aurículo-ventricular
- CAV defecto atrioventricular
- CIA comunicación interauricular
- CIV comunicación interventricular
- CoA coartación aórtica
- EA estenosis aórtica
- EP estenosis pulmonar
- ERAP estenosis ramas pulmonares
- ETP estenosis tronco pulmonar
- IAo insuficiencia aórtica
- MHC miocardiopatía hipertrófica
- PCA ductus arterioso
- ProM prolapso mitral
- TAC Truncus arterioso
- TF Tetralogía de Fallot
Soplos son los ruidos producidos por un flujo turbulento en el corazón y los vasos sanguíneos y/o vibraciones de estructuras cardiacas. Para definirlos bien hemos de referirnos a todas sus características
- Fase del ciclo cardíaco en el que ocurren: sistólico, diastólico, sistodiastólico o continuo. Los soplos sistólicos pueden ser orgánicos o funcionales.
- Duración dentro de la fase del ciclo cardíaco en que sucede el soplo; este puede ser precoz (proto-), medio (meso-), tardío (tele-), u ocupar todo (holo- o pansistólico).
- Morfología o forma en que un soplo pasa de la intensidad mínima a la máxima o viceversa…
- Eyectivo, in crescendo-decrescendo, romboidal o en diamante. Cuando el soplo es creciente hasta alcanzar su intensidad máxima y después decrece progresivamente hasta desaparecer. Es típico de la estenosis de válvulas sigmoideas audible en la base.
- De regurgitación o pansistólico: intensidad constante o rectangular, típico de la comunicación interventricular y de la regurgitación de válvulas auriculoventriculares.
- Intensidad: va del 1 al 6, los poco audibles son un 1 , a partir del 4 se añade un thrill o frémito y los más intensos se advierten sin necesidad del fonendoscopio
- Intensidad: va del 1 al 6, los poco audibles son un 1 , a partir del 4 se añade un thrill o frémito y los más intensos se advierten sin necesidad del fonendoscopio
Denominamos soplo orgánico cuando es un soplo patológico derivado de una anomalía intracardiaca o de los vasos, que provoca un flujo turbulento, hay que diferenciarlo de los soplos inocentes típicos del niño sano.
SOPLOS INOCENTES
Se prefiere la denominación de SOPLO INOCENTE (Evans, 1947) pues deja claro la ausencia de patología. Su etiología no se conoce con seguridad, aunque influye la frecuencia cardiaca más elevada del niño, la mayor elasticidad del miocardio y la menor distancia entre el corazón y el tórax que puede originar que se ausculte el sonido del flujo sanguíneo.
Dado que son los soplos que con mucha más frecuencia va a escuchar el pediatra, es fundamental que los conozca y pueda identificarlos y diferenciarlos de los soplos patológicos.
Sus características son:
- Son de corta duración, nunca holosistólicos. R1 se identificará claramente.
- Intensidad entre grados 1 y 3, por tanto, no presentan frémito.
- La mayoría de ellos, igual que los soplos orgánicos, aumentan de intensidad con la presencia de fiebre, anemia o un gasto cardiaco elevado.
- Tienen una localización definida con escasa irradiación.
- Cambian de intensidad al cambiar la posición del paciente.
El soplo vibratorio de Still es el soplo funcional más frecuente en los niños (75-85% de niños en edad escolar). Puede auscultarse a cualquier edad, desde la lactancia a la adolescencia, pero es más frecuente entre los 2 y los 6 años de edad. Es un soplo de tono medio-bajo, de moderada intensidad (grado 2-3/6), confinado a la primera mitad de la sístole y con máxima auscultación en borde esternal izquierdo y ápex. Se oye mejor en decúbito y a menudo disminuye de intensidad y cambia de tono con los cambios posturales. Lo más característico del soplo de Still es su tonalidad musical o vibratoria.
Otros soplos inocentes son el soplo venoso (en la unión yugular, innominada o subclavia con la vena cava, en el primer espacio intercostal, que desaparece en supino); el soplo de estenosis de ramas pulmonares en el neonato (más audible en axilas, desaparece los primeros meses de vida) y el soplo carotideo de la unión de los vasos supra-aórticos con el arco aórtico (hay que descartar estenosis aórtica)
El diagnóstico diferencial del soplo inocente en el niño asintomático se debe hacer con los soplos patológicos debidos a una comunicación interventricular (CIV), estenosis subaórtica y miocardiopatía hipertrófica, aunque éstos suelen ser más rudos y asociar frémito.
Si quieres saber más sobre soplos cardiacos
En la etapa neonatal, los síntomas y signos clínicos de las cardiopatías congénitas (CC) pueden ser muy variables, dependiendo del tipo de malformación. Hay que tener en cuenta que algunos recién nacidos (RN) pueden no presentar soplo cardíaco al nacimiento, ni mostrar signos clínicos aparentes de malformaciones cardíacas de tipo complejo porque las presiones entre ambos ventrículos están a igual presión al nacimiento. Conforme disminuyen las resistencias pulmonares y/o se cierran las estructuras de la circulación fetal las CC se ponen de manifiesto. En la actualidad, con el diagnóstico prenatal de las cardiopatías se está cambiando el manejo de estos pacientes, tras su diagnóstico el RN nace en hospitales con cardiología y cirugía cardiaca neonatal y pediátrica.
La forma de presentación de los neonatos con algún tipo de cardiopatía sin diagnóstico prenatal, se enmarca dentro de dos formas clínicas: una primera como shock o en forma de cianosis; sin embargo, algunos neonatos tienen formas mixtas, el análisis de esta combinación de presentaciones clínicas permite sospechar el tipo de cardiopatía.
En general casi todas las cardiopatías graves dan síntomas coincidiendo con el cierre del ductus, bien porque éste mantiene el gasto sistémico (al cerrarse provoca shock, porque la aorta descendente se queda sin flujo, son cardiopatías ductus-dependientes del gasto aórtico o sistémico) o el gasto pulmonar (al cerrarse provocará cianosis intensa, cardiopatías ductus-dependiente del gasto pulmonar).
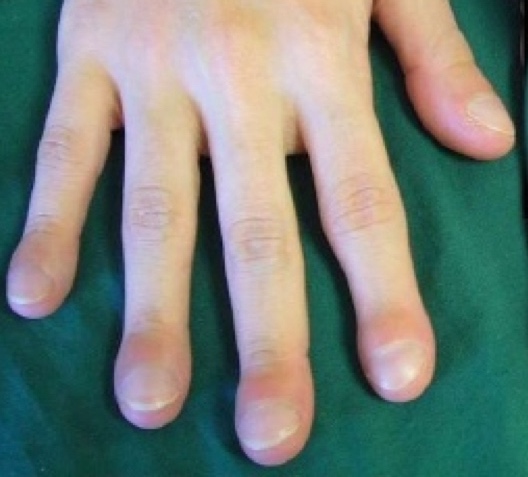
Imagen de acropaquias
En la evolución con los años, el cortocircuito izquierda-derecha de manera mantenida provoca elevación de la presión pulmonar y cambios histológicos del vaso pulmonar generando hipertensión pulmonar, en el tiempo, este aumento del flujo provoca aumento del grosor de la capa muscular media y, consecuentemente, elevación de resistencias pulmonares que llegan a ser irreversibles, es el llamado Síndrome de Einsenmenger, en ese momento el cortocircuito queda invertido de manera irreversible, provocando cianosis (cortocircuito derecha izquierda). Otras complicaciones secundarias al cortocircuito, es el riesgo de endocarditis. En las CC que provocan cianosis existe una producción aumentada de hemoglobina de manera compensatoria, provocando poliglobulia, que son causantes de abscesos como complicación debido la hiperviscosidad sanguínea. Las deformaciones óseas de los dedos (“acropaquias o dedos en palillos de tambor”) son deformaciones de las falanges distales de los dedos de manos y pie secundarias a la hiperproducción sanguínea ósea en esas localizaciones.
En la actualidad el cateterismo intervencionista está solucionando problemas de cardiopatías simples y aisladas y resolviendo las complicaciones que van apareciendo en el seguimiento, condicionando una disminución del número de intervenciones quirúrgicas.
Existen dispositivos específicos para cierre de defectos septales como defectos interauriculares y algunos interventriculares, persistencia de Conducto arterioso o fístulas y se pueden realizar aperturas de vasos estrechos como en la Coartación de aorta o valvuloplastias para las estenosis de válvulas (incluso intervencionismo fetal) y, más recientemente, interposición de stent para apertura de vasos y de válvulas en posición pulmonar y aórtica por cateterismo.
La primera intervención quirúrgica en una cardiopatía congénita se realizó en 1938 en Boston por el Dr Robert Gross y consistió en el cierre de un Conducto arterioso por toracotomía lateral. Link
La aparición en 1958 del oxigenador de membrana supuso un cambio radical en la cirugía, condicionó el poder “parar” el corazón y mantener al cuerpo oxigenado con la circulación extracorpórea (CEC) mientras se realizaba la intervención. Link
El progresivo desarrollo tecnológico, permitió la aparición de la cirugía cardiaca neonatal en los años 80. En 1975, el Dr Jatene intervino con éxito la corrección anatómica de un niño de 1 año con una Transposición de grandes arterias (inversión en la posición de los vasos, de tal manera que la aorta está conectada con el ventrículo derecho y la pulmonar con el izquierdo), técnica que se empezó a utilizar en neonatos por el Dr. Castañeda a partir de finales de los 80.
De entre todas las técnicas, la de más riesgo y la última en desarrollarse ha sido la cirugía que se realiza en los niños afectados de Síndrome de hipoplasia de cavidades izquierdas (SHCI), llamada cirugía Norwood, son recién nacidos que nacen sin desarrollo del ventrículo izquierdo, ni de la aorta, la cirugía consiste en unir la mini aorta y la arteria pulmonar y formar un único vaso, desconectando las ramas pulmonares y manteniendo el flujo a través de un tubo entre ramas pulmonares y ventrículo derecho (o con la aorta dependiendo de la técnica). El desarrollo del diagnóstico prenatal ha disminuido la incidencia del SHCI y los casos que se operan tienen un resultado de supervivencia que varía según el centro entre 60-80%.
Las cirugías cardiacas en cardiopatías congénitas se dividen en cirugías cerradas (sin necesidad de extracorpórea, habitualmente por toracotomía lateral) y abiertas (con CEC). Habitualmente se denominan en función del cirujano que realizó la técnica por primera vez. Si el paciente tiene dos ventrículos de tamaño similar y con buena función, y un vaso de tamaño adecuado, se realiza una cirugía correctora o biventricular (se intentará que el corazón sea lo más parecido al corazón normal), el vaso quedará como aorta y se colocará una prótesis (tubo) en posición pulmonar. Los tubos empleados pueden ser homoinjertos (aorta o pulmonares de cadáveres procesados en un banco de tejidos y desnaturalizados y criopreservados), biológicos (xenoinjerto, de origen animal, o de ingeniería genética) o de material sintético. Todos estos tubos tienen la desventaja de una durabilidad corta y que en el niño pequeño tienen que ser de pequeño tamaño (adecuados a su peso) y deben ser cambiados cuando el niño crece. Cuando no existe posibilidad de realizar una cirugía biventricular por hipodesarrollo o ausencia de un ventrículo o una atresia de una de las válvulas auriculo-ventriculares realizamos una cirugía univentricular (llamada cirugía de Fontan), donde las venas cavas se conectan mediante un tubo (habitualmente de un material protésico de Goretex® o PTLE) con las arterias pulmonares, dejando sólo un ventrículo único como corazón.
Lo que es imprescindible para el éxito de la cirugía en general, es que las ramas pulmonares tengan un desarrollo adecuado, si existe hipoplasia severa de las ramas pulmonares, no se podrá ofrecer una corrección quirúrgica completa.
Otra manera de denominar a las cirugías es clasificarlas en cirugías anatómicas (corrección del defecto según su problema anatómico) o fisiológicas (correcciones quirúrgicas que hacen que el corazón funcione correctamente, aunque no hayamos corregido su defecto anatómico).

Son las más frecuentes, pueden ser defectos a nivel auricular, ventricular o a nivel de vasos
Incluimos: defectos a nivel interauricular (CIA), defectos atrio-ventriculares (denominado Canal AV), defectos septales a nivel interventricular (CIV membranosa si es del septo de la parte alta o CIV muscular cuando son de la parte baja del septo interventricular) y ductus arterioso persistente
Provocan aumento del flujo pulmonar que desencadena insuficiencia cardiaca, el aumento del flujo al circuito pulmonar provoca un disbalance entre el flujo pulmonar y sistémico (Qp/Qs), más sangre va a los pulmones
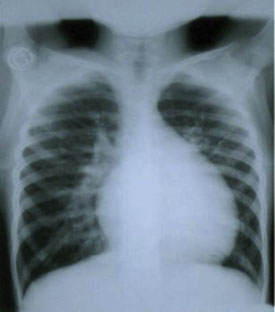
La clínica que produce son soplos y signos de insuficiencia cardiaca y en la radiografía de tórax se observa aumento del flujo pulmonar
El diagnóstico se realiza por Ecocardiografía, raras veces precisa cateterismo cardiaco diagnóstico (sólo para cierre de los defectos, denominado cateterismo intervencionista). En ocasiones se realizan otros estudios de imagen como AngioTAC o AngioRM, sobre todo para ver la anatomía de los vasos que salen del corazón (arterias pulmonares o troncos supraórticos). Además, en todo niño con sospecha de lesión cardiaca se debe realizar ECG para confirmar el ritmo cardiaco, además nos orientará sobre la sobrecarga de las cavidades cardiacas y su repercusión.
La Comunicación interauricular (CIA) es un defecto del tabique interauricular, la más frecuente es la situada a nivel central, CIA tipo Ositum Secundum, hay que diferenciarlo en el niño pequeño de un Foramen oval permeable. Produce paso de sangre de la auricula izquierda a la derecha, provocando sobrecarga sobre las cavidades derechas, más sangre que va al circuito pulmonar, como la zona de las aurículas es de baja presión el soplo se produce por la sangre que pasa a través de la arteria pulmonar, provocando una estenosis pulmonar relativa. Su tratamiento es por cierre por cateterismo cardiaco con diferentes dispositivos en la actualidad, cuando es muy amplia, el niño es pequeño y los bordes del tejido auricular que rodea el defecto es pequeño se plantea la cirugía (mediante la colocación de un parche, se realiza con circulación extracorpórea).
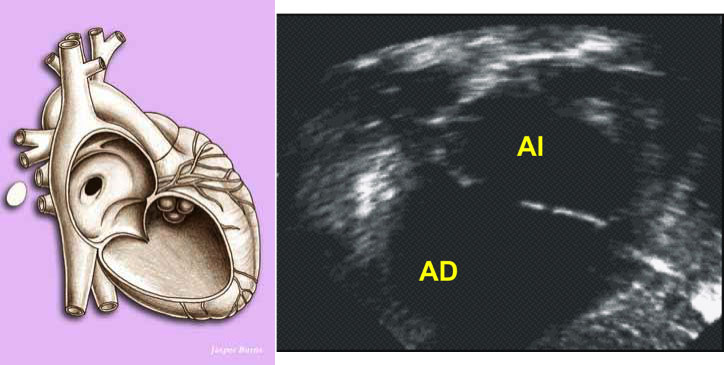
Existen otras CIA en otras localizaciones que no se pueden cerrar por cateterismo, son tributarias de cierre quirúrgico: las situadas en la parte alta del septo llamadas CIA tipo seno venoso, que a veces se acompañan de anomalías de las venas pulmonares que drenan en la aurícula derecha y la CIA tipo Ostium Primum, (defecto de los cojines endocárdicos) localizada en la parte inferior del septo y que suele asociarse a anomalías de la válvula mitral (agujeros a este nivel, que se denominan “cleft”)
La Comunicación interventricular (CIV) más frecuente es la localizada en la parte inferior del septo interventricular, es la llamada CIV muscular, suele cerrarse espontáneamente. Si es muy amplia y requiere cierre se puede hacer por cateterismo mediante la colocación de un dispositivo o rara vez por cirugía cardiaca (con la colocación de un parche). Las CIVs situadas en la parte superior, debajo de los vasos arteriales son las que precisan cierre quirúrgico con mayor incidencia. De momento no se cierra por cateterismo, dado que están muy cerca de la válvula aórtica y pueden dañar la conducción del latido cardiaco, dado que por la zona subaórtica se localiza el haz de Hiss (conducción del latido a los ventrículos) y su daño produce bloqueo aurículo-ventricular. Provocan sobrecarga de las cavidades izquierdas, primero se intenta tratamiento médico y si no se cierran se procede a la cirugía. El flujo aumentado a nivel del pulmón provoca HTP más pronto que las CIA, se debe intervenir antes del año (máximo 2 años). En ocasiones existen CIVs múltiples, amplias e inaccesibles para el cirujano, en esos casos se puede colocar un cerclaje pulmonar (una banda) como cirugía paliativa en espera del cierre de algunas o todas y después se puede proceder a retirar el cerclaje y cierre de las restantes
El ductus arterioso persistente provoca también sobre carga del ventrículo izquierdo si el tamaño es amplio y la cantidad de sangre a su través es elevada. Actualmente el cierre se hace por cateterismo, mediante dispositivos, incluso en la época prenatal. Hay que diferenciar el ductus del recién nacido prematuro, en estos casos se realiza tratamiento médico con paracetamol o ibuprofeno antes del cierre quirúrgico.
El Síndrome de Down se asocia específicamente al defecto atrio-ventricular, su forma más leve es el defecto de la zona baja del septo interauricular (CIA tipo Ostium Primum) hasta su forma más compleja, defecto atrio-ventricular completo con válvula aurículo-ventricular común, requiere cirugía correctora sobre los 6 meses de vida, es característico una alteración ECG con eje izquierdo (por ser un defecto situado en el centro del corazón, donde se encuentra el haz de Hiss, zona de la conducción ventricular). Además, el SDM de Down suele asociarse al ductus arterioso.

Imagen de Defecto atrio-ventricular completo
Son lesiones obstructivas que provocan obstrucción a la entrada o salida de la sangre a uno de los ventrículos. Incluimos:
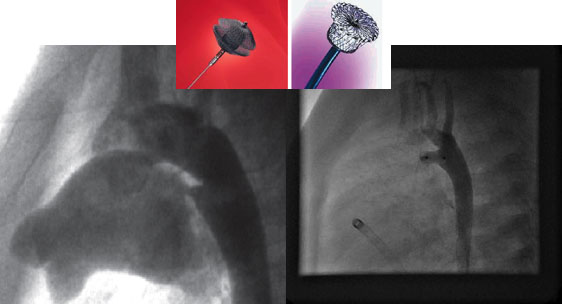
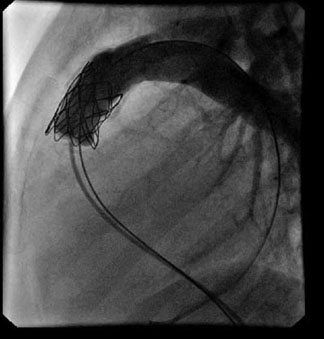
Estenosis pulmonar (si es muy severa y se asocia a un defecto septal puede provocar cianosis por cortocircuito derecha-izquierda), puede ser subvalvular, valvular o supravalvular (muy frecuentemente asociada a síndromes como el SDM Williams). Las estenosis pulmonares valvulares suelen solucionarse mediante cateterismo con una valvuloplastia pulmonar. Habitualmente las subvalvulares y supravalvulares requieren de cirugía correctora. Existen formas graves de hipodesarrollo del ventrículo derecho y de la válvula tricuspidea, denominadas SDM del ventrículo derecho hipoplásico (o Atresia pulmonar con septo integro) el manejo en esos casos es diferente a una lesión única del Tracto de salida del ventrículo derecho
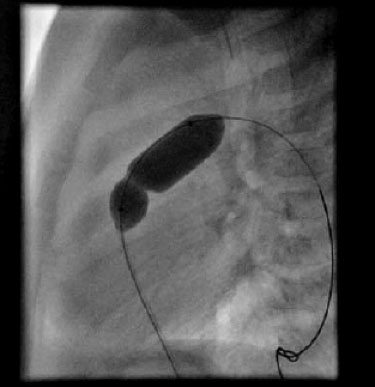
Imagen de una Valvuloplastia pulmonar
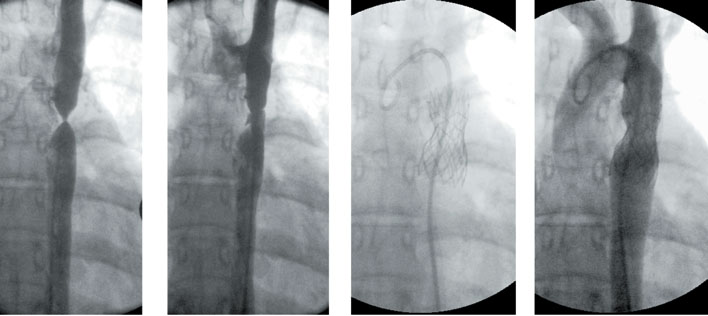
Estenosis aórtica (según el grado de obstrucción se diferencian en ligeras, moderadas o severas). Según la localización de la zona de la estrechez serán subvalvulares (si son localizadas suelen ser debidas a un rodete, puede ser todo el tracto de salida del ventrículo izquierdo englobando el anillo valvular aórtico, lo que complica su tratamiento); valvulares o supravalvulares (característica asociación con el SDM Williams, afecta a la entrada de las coronarias). Su tratamiento verá determinado por el grado de estrechez y localización. En las formas valvulares lo primero es indicar una valvuloplastia aórtica (dilatación con balón por cateterismo, ver siguiente figura) en espera del crecimiento del niño, para colocar una prótesis mecánica cuando sea factible (idealmente tras completar el crecimiento, a partir de los 12 años)
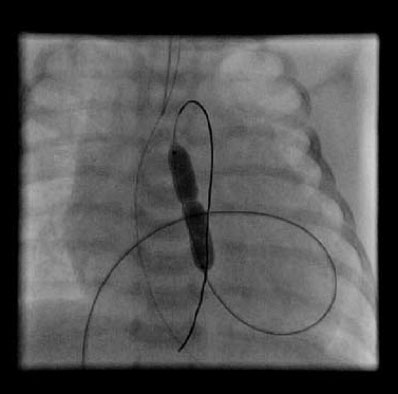
Imagen de una valvuloplastia aórtica
cirugía denominada técnica de Ross o autoinjerto pulmonar en posición aórtica (su válvula pulmonar nativa se coloca en posición aórtica, en la pulmonar se coloca un tubo que requerirá recambio conforme crezca el niño, pero siempre es mejor tolerada la estenosis pulmonar que la aórtica)
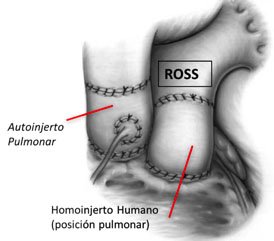
Imagen de la cirugía de Ross
Existen formas de hipodesarrollo del ventrículo izquierdo con lesiones a varios niveles, es característico la asociación de la coartación aórtica con válvula aórtica bicúspide, estenosis aórtica y válvula mitral con un único musculo papilar (válvula en paracaídas) es el denominado Síndrome de Shone y conforme se desarrolla el niño requiere intervenciones a otros niveles además de sobre la válvula aórtica. La forma más grave de las lesiones del lado izquierdo cardiaco es el SDM de hipoplasia de cavidades izquierdas, que se incluyen dentro de las formas de cardiopatías univentriculares.
Coartación aórtica (estrechez tras la salida de la subclavia, en aorta descendente, provoca hipertensión arterial por encima de la obstrucción y disminución de pulsos en la zona inferior a la estrechez. En los recién nacidos y lactantes se trata primero con cirugía y resección de la zona de tejido estrecha, en niños mayores y adultos se puede realizar tratamiento mediante cateterismo (angioplastia dilatación con la colocación de un stent en la zona estrecha).
Otras: Estenosis mitral (rara la forma aislada en niños, suele estar asociada a otras anomalías del corazón izquierdo). También son infrecuentes otras anomalías del corazón izquierdo como las estenosis de venas pulmonares
La cianosis es la coloración azulada de la piel y se manifiesta cuando la concentración de hemoglobina (Hb) reducida en sangre es superior a 5g/dL. Hay que diferenciar cianosis de hipoxia. La hipoxia es una disminución de la concentración de oxígeno en sangre. No todo niño hipóxico está cianótico o a la inversa. Un recién nacido (RN) puede estar hipóxico y no cianótico en caso de anemias graves, puesto que la cantidad de Hb reducida puede ser baja; o puede estar cianótico sin hipoxia, el ejemplo sería en las alteraciones de la hemoglobina, como en las Meta hemoglobinopatías congénitas, el paciente ser verá azul pero el contenido de oxígeno es normal.
En la mayoría de las ocasiones los pacientes con cardiopatías congénitas con SatO2 >65-75% no están hipóxicos, por lo que su respiración es normal (sin polipnea ni dificultad respiratoria) a diferencia de los pacientes con hipoxia secundaria a alteraciones respiratorias. Si la cianosis aparece bruscamente (tras el cierre del ductus) puede provocar polipnea y shock por hipoxia tisular (y elevación de ácido láctico).
Para diferenciar si la causa de una cianosis en el RN es debida a una cardiopatía congénita se realizará un test de hiperoxia, que consiste en la administración de oxígeno al 100% y comprobar si la SatO2 aumenta > 10% o la PaO2 aumenta >20-30 mmHg. En ese caso se trata de patología respiratoria concomitante, si no hay cambios en la SatO2 o PaO2 estaremos ante una cardiopatía cianosante, aunque en ocasiones el RN presenta una situación de persistencia de la circulación fetal (PCF) que es persistencia de la presión pulmonar elevada, típica del feto, lo que condiciona cortocircuito derecha izquierda a través de un resto de Foramen oval o de una ductus arterioso, en estos casos la historia obstétrica nos orienta hacia el diagnóstico.
Una diferencia entre la SatO2 en la extremidad superior derecha y las extremidades inferiores debe hacer sospechar en la PCF (cortocircuito derecha -izquierda a través del ductus arterioso persistente)
Lecturas recomendadas:
Las cardiopatías congénitas cianosantes pueden ser causadas por 2 mecanismos:
- Por obstrucción al corazón derecho asociado a un defecto que permite un cortocircuito derecha-izquierda a su través: como la Atresia pulmonar con CIV, la Atresia pulmonar con septo íntegro (con cortocircuito derecha-izquierda a través del agujero oval), o Atresia tricuspídea o Ventrículo único con Estenosis pulmonar. En estos casos el flujo pulmonar estará disminuido porque hay un porcentaje de sangre que no pasa al circuito pulmonar, se dirige directamente a la aorta (circulación sistémica) (Flujo pulmonar/ sistémico o Qp/ Qs inferior a 1).
- Por circulación en paralelo como en la d-Transposición de grandes arterias, donde hay un cambio en la disposición de los vasos, provocando que la sangre que se dirige al circuito pulmonar no va al circuito sistémico y se requiere de una comunicación para que la sangre oxigenada llegue a la aorta. Habitualmente tras el cierre del ductus arterioso aparece una cianosis intensa. La cantidad de sangre que va al circuito pulmonar y sistémico es casi igual, pero en paralelo
Existe otro concepto importante en las CC y es el de la mezcla. Pueden existir CC con cortocircuito derecha-izquierda, pero sin cianosis porque la mezcla a nivel intracardiaco es elevada y la cantidad de sangre que llega al circuito pulmonar es adecuada (Ej. un Ventrículo único sin obstrucción al flujo pulmonar o un defecto atrio-ventricular) o bien, cuando el grado de estrechez del tracto de salida del VD permite un paso de sangre suficiente al circuito pulmonar, estando el paciente “balanceado” (sin hipoxia ni cianosis severa).
En los casos de obstrucción pulmonar grave sin cortocircuito derecha-izquierda el corazón intentará compensar la obstrucción y si no lo consigue provocará shock, al no llegar la sangre al circuito sistémico.
Es la más común de las cardiopatías que cursan con cianosis. Representa el 5-8% de todas las cardiopatías congénitas. Incluye un amplio espectro, la forma clásica, de la cual se tratará fundamentalmente en este apartado, dejando para el final algunas características especiales de la atresia pulmonar con CIV y la tetralogía de Fallot (TF) con ausencia de la válvula pulmonar.
La microdeleción 22q11 y la trisomía 21 se asocian a la TF. Del 15 al 25% de los pacientes con Fallot presentan la microdeleción 22q11, también conocida como síndrome CATCH 22 (defectos cardiacos, anomalías faciales, hipoplasia tímica, hendidura palatina e hipocalcemia).
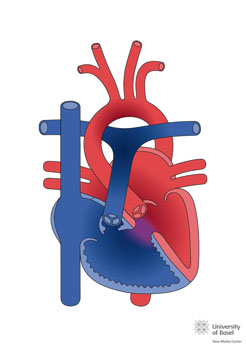
Los cuatro elementos básicos en la anatomía son:
- Desviación anterior y craneal del septo infundibular, lo que ocasiona estenosis a dicho nivel, acompañada en mayor o medida de hipoplasia de válvula, tronco y ramas pulmonares. Lo que marca la mayor severidad y peor pronóstico en ésta cardiopatía es el grado de hipoplasia a este nivel.
- Comunicación interventricular (CIV) subaórtica amplia.
- Aorta cabalgando sobre la CIV por lo que se relaciona con ambos ventrículos.
- Hipertrofia ventricular derecha, secundaria a la obstrucción en el tracto de salida ventricular derecho.
FISIOLOGIA
La comunicación interventricular es amplia, por lo que la dirección y magnitud del cortocircuito a su través dependerá del grado de estenosis pulmonar, que además tiende a ser progresiva. Es frecuente que inicialmente la estenosis sea ligera con lo que predomina el cortocircuito izquierda-derecha, en ocasiones, con síntomas de insuficiencia cardiaca, conocido como Fallot rosado. A medida que progresa la enfermedad y la estenosis se hace más severa se invierte el cortocircuito y aparece cianosis, situación más frecuente.
La clínica varía en función del grado de estenosis pulmonar, el 25% tienen cianosis al nacimiento. En el niño mayor con TF no corregida y cianosis severa es característica la postura de “cuclillas” que favorece el cortocircuito izquierda-derecha por la comunicación interventricular incrementando el flujo pulmonar y mejorando la hipoxemia.
Las crisis hipoxémicas son episodios bruscos de cianosis, desencadenados por llanto, alimentación o defecación, acompañados de polipnea y pérdida de conocimiento. Las crisis hipoxémicas también pueden aparecer ante ciertas situaciones como fiebre o ejercicio intenso y se deben a una disminución crítica del paso de sangre por el infundíbulo pulmonar siendo característica la desaparición del soplo eyectivo. Pueden aparecer en niños con o sin cianosis previa. Es un signo clínico de gravedad e implica cirugía urgente.
A la exploración predomina la cianosis, aunque inicialmente puede estar ausente o ser muy ligera. En la auscultación cardiaca destaca un soplo sistólico en borde esternal izquierdo superior, es tanto más holosistólico cuanto menos severa es la estenosis. Típicamente este soplo disminuye o desaparece durante las crisis hipoxémicas. El componente pulmonar del segundo ruido está ausente o es muy débil.
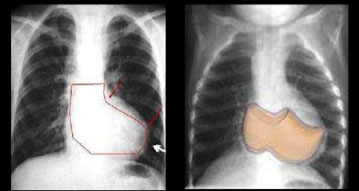
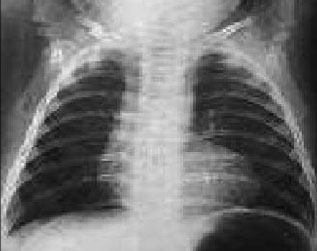
En la Rx de tórax la silueta cardiaca normal o pequeña con levantamiento de la punta cardiaca y cono de arteria pulmonar disminuido (corazón en zueco, ver siguiente imagen). La vascularización pulmonar es normal o se encuentra disminuida.
La Ecocardiografia es el método diagnóstico de elección y en la mayor parte de los casos aporta la información necesaria para la corrección quirúrgica. Permite visualizar la CIV subaórtica, las características del cortocircuito a su través, el porcentaje de cabalgamiento aórtico, el grado y localización de la estenosis pulmonar, el tamaño de anillo, tronco y ramas pulmonares principales y la existencia de lesiones asociadas.
TRATAMIENTO
Excepcionalmente pueden precisar tratamiento en el periodo neonatal, en recién nacidos con estenosis pulmonar severa que presentan cianosis con hipoxemia al cerrarse el ductus, en estos casos es obligatorio el uso de prostaglandinas y cirugía precoz. La mayoría de los pacientes no precisan tratamiento médico en espera de la cirugía aunque en ocasiones se utilizan betabloqueantes, con objeto de relajar el infundíbulo pulmonar y prevenir las crisis hipoxémicas.
Las crisis hipoxémicas son una urgencia médica, cuyo tratamiento consiste en oxigenoterapia, posición genu-pectoral, administración rápida de volumen, cloruro mórfico, y si es muy severa vasopresores (ideal el uso de fenilefrina que es un agonista alfa puro, sin efecto beta adrenérgico, que puede aumentar la crisis) o esmolol intravenoso (betabloqueante de acción ultracorta). Su prevención consiste en evitar la irritabilidad del niño, incluyendo la administración de sedantes, y evitar el estreñimiento. La cianosis severa y/o progresiva y la presencia de crisis hipoxémicas son indicación de tratamiento quirúrgico urgente.
La edad habitual de corrección electiva es entre los 3 y 6 meses. Consiste en cerrar la comunicación interventricular con un parche y corregir la estenosis pulmonar con diversas técnicas. La más frecuente es la apertura del infundíbulo y del anillo pulmonar mediante un parche que se extiende hasta el origen de una rama pulmonar (parche transanular). El precio a pagar por ello es la aparición progresiva de insuficiencia pulmonar con dilatación y disfunción del ventrículo derecho en el futuro. La tendencia actual es a preservar en lo posible la función de la válvula pulmonar.
En ocasiones por la edad o anatomía desfavorable no se puede realizar de primera intención la corrección completa, siendo necesarios procedimientos paliativos, como fístula de Blalock-Taussig modificado (interposición de un tubo de 4-5mm entre la aorta o subclavia con la arteria pulmonar mediante cirugía) o colocación de un stent en el ductus a través de un cateterismo en el recién nacido.
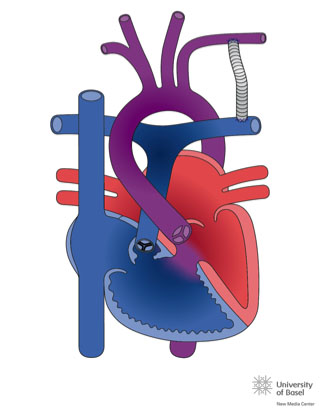
Imagen de Fistula BT izquierda modificada. Tomada de http://www.chd-diagrams.com/heart-operation/155
El pronóstico a largo plazo de la TF clásica es favorable, la supervivencia global es del 90% a 30 años, aunque es frecuente la necesidad de nuevas intervenciones quirúrgicas o procedimientos intervencionistas en las ramas pulmonares. A largo plazo el mayor problema es la insuficiencia pulmonar, consecuencia del parche transanular, cuyo tratamiento es la implantación de una prótesis en posición pulmonar mediante cirugía o más recientemente de forma percutánea.
Se considera la forma más severa del espectro del Fallot, aunque embriológicamente puede tener otro origen. Desde el punto de vista anatómico la anatomía intracardiaca es similar con CIV subaórtica amplia, cabalgamiento aórtico, y atresia del tracto de salida del ventrículo derecho. La complejidad diagnóstica y terapéutica de esta entidad lo marca la circulación pulmonar.
El mejor caso es en el que se han formado normalmente las ramas pulmonares y están suplidas por un ductus. En la zona peor del espectro las ramas pulmonares centrales no existen o son sumamente hipoplásicas y la circulación pulmonar procede de ramas colaterales sistémico-pulmonares, coronarias o de la circulación bronquial
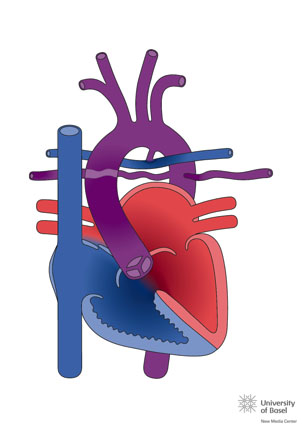
En las formas favorables el tratamiento consiste en la estabilización del flujo pulmonar en el periodo neonatal mediante una fístula de BT o la implantación de un stent en el ductus. En un segundo tiempo el tratamiento definitivo es en el cierre de la CIV y la interposición de un conducto valvulado entre el VD y las arterias pulmonares (técnica de Rastelli). Estos pacientes deben ser reoperados varias veces para recambiar los conductos, la duración media de los mismos durante la infancia varía según el tipo utilizado, con una media alrededor de los 4 años.
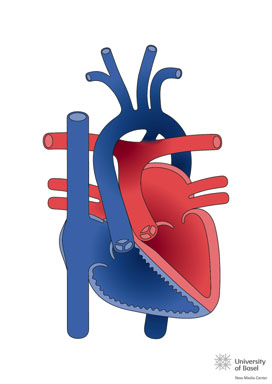
La transposición de grandes arterias (TGA) también descrita como transposición completa de grandes arterias, es una malformación cardiaca congénita caracterizada por concordancia atrio-ventricular y discordancia ventrículo-arterial. En la TGA la aurícula morfológicamente derecha conecta con el ventrículo morfológicamente derecho, del cual se origina la aorta (enteramente o de forma predominante); y la aurícula morfológicamente izquierda conecta con el ventrículo morfológicamente izquierdo, del cual se origina el tronco pulmonar (enteramente o de forma predominante).
La anomalía fisiológica predominante en la TGA es que el retorno venoso sistémico recircula al territorio sistémico a través del ventrículo derecho (VD) y la aorta, mientras que el retorno venoso pulmonar recircula a los pulmones a través del ventrículo izquierdo (VI) y la arteria pulmonar. Por lo que, mientras en el corazón normal las circulaciones sistémica y pulmonar se conectan en serie, en la TGA funcionan como dos circuitos separados, en paralelo.
La presencia de cianosis en el primer día de vida sugiere el diagnóstico de TGA, y si el septo interventricular está íntegro la cianosis puede ser severa ya en las primeras horas de vida. Cuando existe obstrucción en la vía de salida pulmonar, la cianosis es más prominente, mientras que puede ser menos aparente e incluso puede estar ausente en pacientes con CIV amplia (en estos casos predominan los signos de insuficiencia cardiaca congestiva). De forma habitual el RN con TGA se presenta visiblemente cianótico y con una discreta taquipnea, sin dificultad respiratoria franca. La cianosis no varía con el llanto o la administración de oxígeno. No se suelen auscultar soplos a menos que exista obstrucción en tractos de salida. Tras las primeras dos semanas de vida, en pacientes no tratados, suele aparecer falta de medro en relación con la disminución de la ingesta y el aumento de los requerimientos calóricos que supone el incremento del trabajo respiratorio.
La radiografía de tórax y el ECG pueden ayudar en la fase de diagnóstico, pero sus hallazgos son inespecíficos. El ECG muestra habitualmente el predominio derecho normal y en la RX se puede apreciar una ligera cardiomegalia con el pedículo vascular estrecho (silueta con forma ovoide) con circulación pulmonar conservada.
La ecocardiografía es el método de imagen definitivo en el diagnóstico, proporcionando una precisa información sobre los detalles anatómicos y funcionales de la cardiopatía.
El objetivo inicial en el manejo es asegurar una aceptable mezcla circulatoria. La presencia de un defecto septal auricular o ventricular que proporcione una mezcla satisfactoria con una adecuada oxigenación arterial, permite planear o posponer una cirugía correctiva posterior sin la necesidad previa de procedimientos paliativos.
Se administra inicialmente una infusión intravenosa de prostaglandina E1 para mantener la el ductus permeable y mejorar la oxigenación (dosis: 0,01-0,1 microg/kg/min), y es esencial el mantenimiento del equilibrio ácido-base, temperatura y la corrección de la hipoglucemia. En los RN severamente cianóticos o acidóticos tras la perfusión de prostaglandina E1 deben realizarse la septostomía de Rashkind para la estabilización, en la actualidad se realiza en la misma unidad neonatal, bajo control ecocardiográfico (en algunos hospitales se sigue realizando en la Unidad de hemodinámica bajo control por escopia, pero requiere traslado del recién nacido habitualmente inestable hemodinámicamente). Consiste en la introducción de un catéter específico que tiene un balón que se infla en el lado auricular izquierdo y se estira provocando una rotura del septo de la fosa oval, creando una comunicación interauricular.
La operación de intercambio arterial o Switch arterial (técnica de Jatene, es el procedimiento de elección para la corrección quirúrgica de esta cardiopatía en la actualidad.
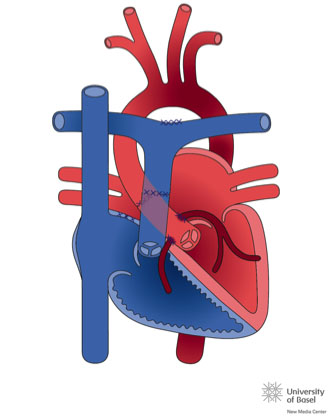
Cirugía de Jatene. Imagen de http://www.chd-diagrams.com/heart-operation/171
Se realiza de forma preferente antes de las dos primeras semanas de vida. La intervención consiste en seccionar la aorta y la arteria pulmonar justo por encima de sus senos de Valsalva, y re-anastomosándolas en sus correctas posiciones anatómicas, pasando la aorta ascendente a una posición posterior con respecto a las arterias pulmonares (maniobra de Lecompte). Las arterias coronarias son removidas desde su antigua raíz junto con un botón de pared aórtica y reimplantadas en la antigua raíz pulmonar. Este tipo de corrección tiene una supervivencia del 90-95% en las TGA no complicadas, restablece las relaciones fisiológicas normales de los flujos arteriales sistémico y pulmonar.
En las técnicas de reparación quirúrgica tipo Switch auricular (procedimientos de Mustard o de Senning, el retorno venoso sistémico es redirigido a nivel auricular al VI, y por otra parte el retorno venoso pulmonar hacia el VD. Es una técnica que se empleó hace años, queda relegada a escasos pacientes con especial anatomía.
Cirugía de Senning. Imagen de http://www.chd-diagrams.com/heart-operation/172/
Cuando existe obstrucción a nivel el tracto de salida del VI, puede ser necesario interponer un conducto extracardiaco entre el VI y el tronco pulmonar.
Todos los pacientes intervenidos requieren un seguimiento de por vida. En los que se realizó una técnica de Switch auricular, son frecuentes a partir de la segunda-tercera década de la vida la aparición de arritmias y signos de disfunción del ventrículo sistémico, morfológicamente derecho. Los pacientes sometidos a una técnica de Switch arterial pueden presentar problemas en relación con la aparición de estenosis coronarias, dilatación de la raíz aórtica e insuficiencia aórtica.
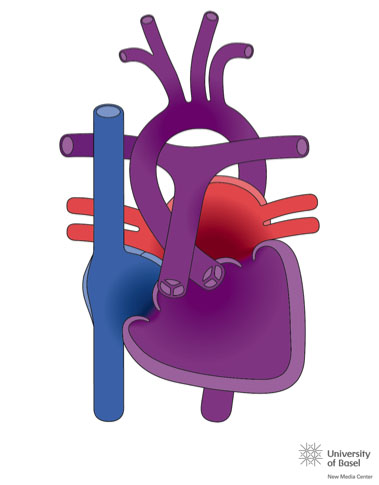
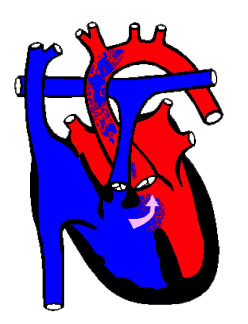
El hecho fundamental de las anomalías cardiacas complejas agrupadas bajo el término de corazón univentricular es que la conexión atrioventricular es univentricular, es decir, se realiza completa o predominantemente a una sola cámara ventricular. La mayoría de estos corazones poseen dos ventrículos, uno grande (ventrículo dominante) y otro pequeño en el que prácticamente siempre falta su cámara de entrada (ventrículo incompleto, hipoplásico, pequeño). El ventrículo único, aunque funcionalmente sólo presenta una cámara funcional, existe un resto “bulbo-ventricular” que es el resto del otro ventrículo no desarrollado. Cuando existe obstrucción al flujo pulmonar aparecerá cianosis, si el flujo pulmonar está conservado, dada la mezcla intracardiaca, puede no existir cianosis dado que el flujo pulmonar estará aumentado.
Al faltar un ventrículo o una válvula aurículo-ventricular la corrección nunca puede establecerse biventricular. Se realizará cirugía univentricular o de Fontan, ésta consiste primero en la conexión de la vena cava superior a la rama pulmonar derecha, entre los 4 meses y 1 año es la cirugía de Glenn http://www.chd-diagrams.com/heart-operation/235 y, posteriormente, alrededor de los 4 años, cuando se desarrolle el hemicuerpo inferior, la vena cava inferior se conecta a la rama pulmonar. Existen muchas técnicas del Fontan, al inicio se realizaba conectando directamente la aurícula derecha con las ramas pulmonares. En la actualidad la técnica más habitual es el Fontan extracardiaco, consiste en la interposición de un tubo de Goretex® o PLTE entre la vena cava inferior y la rama derecha pulmonar (ver imagen)
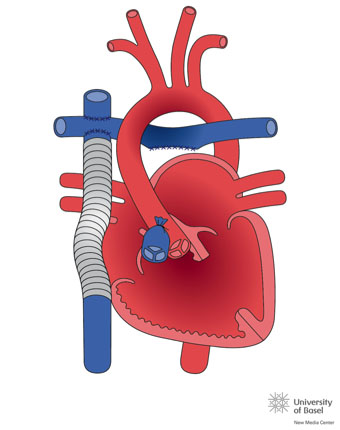
En esta cirugía no existe ventrículo derecho, la sangre venosa alcanzará la circulación pulmonar, quedando una circulación única. El paciente dejará de estar cianótico, aunque una pequeña cantidad de la sangre venosa coronaria drenará en la aurícula única. Los problemas en el seguimiento vienen derivados de la ausencia del ventrículo derecho y su pulsatilidad, con aparición de estasis hepático retrógrado y consecuentemente cirrosis hepática, lesión del flujo mesentérico con aparición de una SDM de malabsorción intestinal, denominado Enteropatía pierde-proteínas, junto con el deterioro del ventrículo único, en ocasiones morfológicamente derecho, lo que provoca fallo cardiaco, precisando de un trasplante cardiaco.
La hipertensión pulmonar (HTP) se define como un aumento en la presión arterial pulmonar (PAP) media > 25 mmHg medida en el cateterismo cardiaco, y puede ser producida por diversos mecanismos etiopatogénicos como señala la clasificación de la Hipertensión pulmonar (Dana Point 2008; Niza 2013; última actualización Niza 2018):
- Hipertensión arterial pulmonar (HAP), producida por obstrucción progresiva del lecho arteriolar pulmonar (fenómenos de proliferación celular, inflamación y trombosis en las arteriolas pulmonares)
- HTP venocapilar (elevación de presión en Aurícula izquierda) por enfermedad del corazón izquierdo.
- HTP secundaria a hipoxemia y/o enfermedad pulmonar.
- HTP por obstrucción tromboembólica del lecho arterial pulmonar crónica.
- HTP por enfermedades de la vasculatura pulmonar.
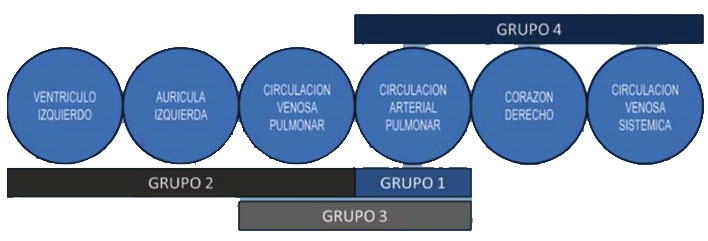
Modificado de Saavedra LE y col. Avances Cardiol 2009;29(2):165-178
Las CC van a producir HTP por las causas I y II fundamentalmente. En la actualidad existen múltiples tratamientos farmacológicos dirigidos a evitar la vasoconstricción pulmonar y el remodelamiento del vaso pulmonar, cuyas vías de acción son diversos: fármacos que actúan sobre las prostaciclinas, antagonistas de los receptores de la endotelina y provocando el aumento de GMPc (inhibiendo la fostodiesterasa tipo 5), todos ellos tienen un efecto vasodilatador pulmonar. La HTP si no se controla va evolucionando y provoca una hipertrofia del vaso pulmonar que genera fallo ventricular derecho y muerte del paciente. Cuando la progresión es muy severa y con el tratamiento médico no se estabilizada se debe someter el paciente a un trasplante pulmonar. Link
Existen un número, cada vez más pequeño, de pacientes que no pudieron ser intervenidos en edad pediátrica, o bien, su clínica pasó desapercibida y desarrollaron en el seguimiento elevación de la presión pulmonar que condicionó el Síndrome de Einsenmenger. Éste consiste en una hiperplasia del músculo del vaso pulmonar, generando elevación fija de las resistencias pulmonares. Si la cardiopatía subyacente es un defecto septal, el cortocircuito izquierda-derecha se invierte, pasando a ser de derecha-izquierda, con lo que el paciente empieza a estar cianótico (coloración azulada de la piel por aumento de la cantidad de hemoglobina reducida en sangre) e incluso puede llegar a estar hipóxico (falta de oxígeno en los tejidos). La hipoxia condiciona aumento de la producción de eritrocitos a nivel medular, consecuentemente aparece poliglobulia. Según los niveles de hemoglobina, cuando ésta es elevada aparece un aumento de la viscosidad de la sangre que puede condicionar aparición de complicaciones como fenómenos embólicos y abscesos.
En situación de Eisenmenger los pacientes no pueden ser intervenidos de su cardiopatía de base. Es muy importante evitar hipotensión, anemia y otros factores ambientales que pueden desencadenar crisis de hipertensión pulmonar y muerte. Sólo el trasplante cardiopulmonar, o el trasplante pulmonar con corrección de su cardiopatía, si ésta es simple, son las opciones de tratamiento.
En la actualidad con los nuevos fármacos que controlan la presión pulmonar la supervivencia de los pacientes de Eisenmenger es cada vez mayor. La tasa de sobrevida es 77% a los 15 años y 42% a los 25 y las causas de muerte más comunes son muerte súbita (30%), insuficiencia cardíaca (25%), y hemoptisis (15%).
La endocarditis infecciosa (EI) es un proceso inflamatorio de etiología infecciosa que afecta al endocardio y/o las válvulas cardiacas. La superficie endotelial se lesiona inicialmente por el flujo turbulento en niños con cardiopatías congénitas o por lesión directa de catéteres centrales o bacteriemias de otro origen en niños sin cardiopatía. Sobre el endotelio dañado se deposita fibrina, plaquetas y en ocasiones hematíes, formando un trombo aséptico. Los patógenos más comunes asociados con la EI en los niños son los Estreptococos del grupo Viridans y el Estafilococo (S.) aureus. La presentación depende de la enfermedad cardiaca de base, el grado de afectación de otros órganos (embolización) y el agente causal.
El diagnóstico se basa en la historia clínica, la exploración física, hemocultivos y otras pruebas de laboratorio y el ecocardiograma. La EI tiene una presentación clínica muy variable y existen criterios diagnósticos (Criterios de Duke) que deben interpretarse como una guía clínica para ayudar al diagnóstico.
Es importante conocer la profilaxis de la EI en niños con cardiopatías, así como los criterios para el diagnóstico de Endocarditis.
Lectura recomendada:
La miocarditis es un proceso inflamatorio del miocardio, su incidencia es desconocida puesto que puede cursar sin síntomas, y causar en la evolución miocardiopatía dilatada crónica. Puede tener diferentes formas de presentación, casos asintomáticos, una forma similar al síndrome coronario agudo que se manifiesta con dolor precordial y una forma severa con disfunción cardiaca e incluso de shock cardiogénico, que en ocasiones, requieren de asistencia ventricular como ECMO, o incluso trasplante cardiaco si no hay recuperación ni respuesta terapéutica.
La sospecha clínica, la elevación de las troponinas y la ecocardiografía ayudan al diagnóstico. Cada vez más tiene un papel importante en el diagnóstico la RM cardiaca, que puede corroborar la sospecha diagnóstica al detectar signos de edema y de inflamación, aunque la prueba más específica es la Biopsia endomiocárdica (BEM). En la actualidad la BEM se realiza cuando el paciente no evoluciona adecuadamente, y puede ayudar al tratamiento. Si existe resto del virus en el tejido cardiaco algunos centros tratan con Interferón, y si existen signos de inflamación sin virus se trata con inmunosupresores (corticoides y/o micofenolato).
Se desconoce por qué algunos niños desarrollan miocarditis y evolucionan a miocardiopatía dilatada, e incluso pueden hacer recaídas. Existen datos de alteraciones inmunológicas, por lo que la administración de Gammaglobulinas se añade al tratamiento de estos niños en el momento del diagnóstico.
Para más información recomendamos la lectura de Protocolos UPIIP: Miocarditis aguda en pediatría, 2016.
La pericarditis es la inflamación de pericardio, las causas de pericarditis se pueden dividir en infecciosas y no infecciosas. Entre el 40 y 86% de los casos se consideran de etiología idiopática, aunque se cree que muchas de ellas son secundarias a una infección vírica. La inflamación del pericardio está causada por una infiltración de granulocitos y linfocitos. Más tarde se puede producir vasodilatación local, aumento de permeabilidad capilar y masiva aparición de proteínas y líquido en el espacio pericárdico (derrame).
La patogenia de los signos y síntomas del derrame pericárdico está determinada por el aumento de la presión intrapericárdica, y depende no sólo de la cantidad absoluta de líquido, sino también de la rapidez con que se acumula y de las características del propio líquido pericárdico. La acumulación rápida se tolera peor que la lenta, que puede permitir un gran acúmulo de líquido sin producir síntomas
Los cambios en el ECG asociados a enfermedad pericárdica son la consecuencia del propio pericardio enfermo y del miocardio subyacente también inflamado o comprimido.
La pericarditis es la causa más común de elevación del ST en el niño. Se definen varias fases en la evolución. En la inicial hay elevación generalizada del segmento ST. Esta fase puede durar horas o pocos días, y no es constante.
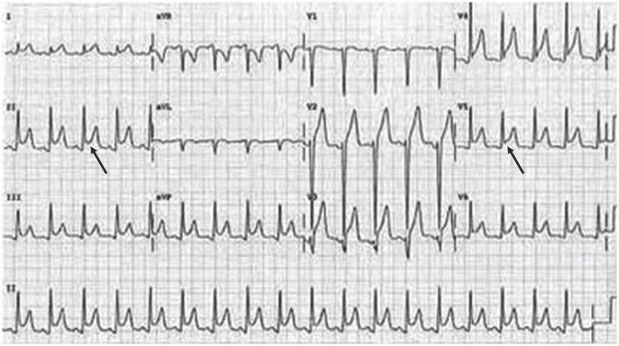
Se sigue de aplanamiento del ST y de la onda T. Posteriormente se invierte la onda T de forma difusa, pudiendo durar esta fase hasta semanas o meses, y finalmente se normaliza la onda T. En los casos de derrame pericárdico importante, son frecuentes los voltajes bajos.
El diagnóstico se hace además de por la sospecha clínica (fiebre con dolor precordial que se exacerba con el supino y la auscultación en ocasiones de roce pericárdico), con la Rx de tórax que muestra cardiomegalia y la ecocardiografía que suele ser la mejor herramienta diagnóstica, pues, confirma la existencia de líquido en cantidad anormal en la cavidad pericárdica.
El tratamiento dependerá del agente causal y de la cantidad de líquido. El tratamiento de la pericarditis es principalmente médico y se basa en reposo y antiinflamatorios, además del tratamiento etiológico, si lo hubiera. En el caso de las pericarditis purulentas el derrame espeso que causa requiere frecuentemente de un procedimiento invasivo para su drenaje. En la pericarditis tuberculosa a pesar del tratamiento antituberculostático, el derrame que causa es difícil de drenar mediante pericardiocentesis. La pericardiectomía debe considerarse prioritaria.
La Enfermedad de Kawasaki (EK) es una vasculitis aguda autolimitada de origen desconocido, propia de la infancia, que afecta sobre todo a las arterias coronarias. La enfermedad es mucho más frecuente en Asia; la incidencia en Europa occidental es inferior a 10 casos por 100.000 habitantes y año, mientras que en Japón es hasta 20 veces superior. Existe cierto patrón estacional, con un aumento del número de casos en invierno y primavera; ocasionalmente hay picos de incidencia parecidos a ciclos epidémicos. La mayor parte de casos ocurren entre los 3 meses y los 5 años de edad. La causa de la EK es desconocida, pero los datos clínicos y epidemiológicos son muy sugestivos de un origen infeccioso.
En la fase aguda se desencadena una inflamación sistémica con aumento de linfocitos, sobre todo B, y activación de la cascada de las citoquinas, que generan una panvasculitis que afecta predominantemente a arterias de pequeño y mediano calibre. En los vasos más afectados se destruye la lámina elástica, lo que permite la dilatación, conduciendo a la formación de aneurismas y aparece una fibrosis progresiva que genera estenosis de los vasos afectos.
El diagnóstico se basa en la existencia de fiebre al menos 5 días y al menos 4 de los 5 restantes criterios clínicos:
- Inyección conjuntival bilateral, no exudativa.
- Alteraciones de labio o mucosa orofaríngea: enrojecimiento intenso y fisuras labiales , exantema orofaríngeo de intensidad variable,l engua de aspecto de fresa.
- Exantema polimorfo (morbiliforme, o semejante al de rubéola o escarlatina), que compromete principalmente tronco y extremidades.
- Edema duro de manos y pies, acompañado o no de enrojecimiento de palmas y plantas. Descamación periungueal en la segunda o tercera semana.
- Linfadenopatía cervical, generalmente única, de más de 1,5 cm de diámetro.
En presencia de fiebre y alteraciones coronarias puede hacerse el diagnóstico con menos de 4 criterios principales. Hasta un 20% de pacientes no reúne todos los criterios principales se conocen como EK incompletas y afectan especialmente a niños menores de un año. En estos pacientes el diagnóstico puede demorarse y, por tanto, la probabilidad de desarrollar complicaciones coronarias es mayor.
NOTA: La EK debe considerarse como diagnóstico en lactantes menores de 6 meses de edad con fiebre prolongada de origen no aclarado y datos de laboratorio de inflamación sistémica.
Las lesiones coronarias aparecen entre 1 y 4 semanas tras el inicio de la fiebre en alrededor del 20% de pacientes no tratados y son responsables de la mayor parte de la mortalidad y morbilidad de la EK. Pueden detectarse dilataciones coronarias de tamaño variable, que afectan fundamentalmente a la zona proximal de las coronarias principales. Pueden coexistir con zonas de estenosis y son susceptibles de aparición de trombos y producir infartos de miocardio.
El ecocardiograma, por su disponibilidad y su alta sensibilidad y especificidad, es la técnica de elección para detectar las alteraciones coronarias. Debe realizarse al diagnóstico, a las 2-4 semanas y de forma periódica para seguimiento de los pacientes con aneurismas. Otras técnicas de imagen, como la resonancia magnética o la tomografía multicorte, pueden ser útiles para detectar aneurismas distales o definir complicaciones como trombosis u oclusión. La coronariografía convencional proporciona mejor definición de las lesiones, pero no debe realizarse hasta la completa resolución del proceso inflamatorio.
El Tratamiento de la fase aguda se realiza con inmunoglobulina G intravenosa (IGGIV), 2 g/kg en una dosis única preferiblemente antes del 10º día de enfermedad, produce, por un mecanismo no bien conocido, una disminución rápida de la actividad. El ácido acetilsalicílico (AAS) se utiliza asociado a la IGGIV para disminuir la sintomatología y detener la actividad inflamatoria. Inicialmente se administran dosis antiinflamatorias, 80-100 mg/kg/24 h en 4 dosis, y cuando el paciente está afebril durante 48-72 h se disminuye la dosis a 3-5 mg/kg/24 h para mantener su acción como antiagregante plaquetario.
En los pacientes sin evidencia de lesión coronaria se mantiene tratamiento antiagregante plaquetario con AAS a dosis bajas durante 6-8 semanas, hasta que la VSG se normaliza y el ecocardiograma confirma la ausencia de anomalías coronarias.
En pacientes con aneurismas pequeños, el tratamiento antiagregante se mantiene hasta que se demuestra ecocardiográficamente la resolución de los mismos. En presencia de aneurismas grandes o muy numerosos debe añadirse anticoagulación o, en niños pequeños, heparina de bajo peso molecular. El 50% de los aneurismas regresan espontáneamente en un plazo de 1-2 años, sobre todo en niños menores de un año y cuando son de pequeño tamaño. Sin embargo, pueden quedar engrosamientos vasculares y aumento de rigidez arterial que actúan como un incremento de riesgo cardiovascular en la vida adulta.
En niños mayores con lesiones coronarias, las recomendaciones sobre la práctica de deporte dependen de la situación clínica y de las pruebas de perfusión miocárdica.
La Electrocardiografía (ECG) es una técnica que consiste en el registro gráfico de la actividad eléctrica que se genera en el corazón. Aporta datos sobre función cardiaca, trastornos del ritmo y de la conducción, hipertrofia de cavidades y ayuda al diagnóstico de cardiopatías congénitas o adquiridas. Su normalidad no descarta afectación cardiaca.
En el niño hay que tener en cuenta las características diferentes al del adulto, sobre todo derivado de los cambios del peso y talla, frecuencia cardiaca (los intervalos PR y Qt varían con la edad) y del cambio que se produce desde el nacimiento a la edad adulta, donde inicialmente el ventrículo derecho es el predominante y hasta los 12-14 años existen datos de hipertrofia del ventrículo derecho que son considerados normal edad. La frecuencia cardiaca disminuye con la edad, mientras que la duración de la onda P, del complejo QRS y el intervalo PR aumentan.
Para la correcta interpretación del ECG, es básico conocer las derivaciones, su orientación espacial y su polaridad. El ECG se basa en el registro gráfico del impulso eléctrico (vector) que genera una onda, la cual puede ser positiva (+) si se acercan al polo positivo de la derivación, y una onda negativa (-) si se aleja del mismo. El ECG de superficie habitual consta de 12 derivaciones, 6 derivaciones frontales y 6 horizontales (o precordiales).
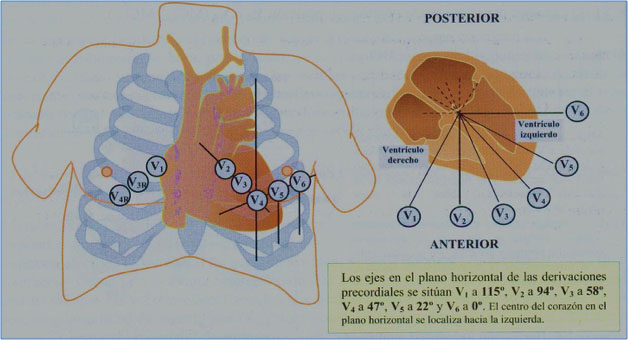
Requiere una lectura sistemática siguiendo una secuencia para obtener un estudio completo. La siguiente secuencia es una de las muchas que se pueden utilizar:
- Ritmo (sinusal o no).
- Frecuencia cardiaca.
- Eje QRS.
- Intervalos: intervalo PR, duración QRS, intervalo QT.
- Segmento ST y onda T.
- Estudio de crecimientos de cavidades.
Dada toda la información existente en la actualidad recomendamos la lectura de los siguientes recursos:
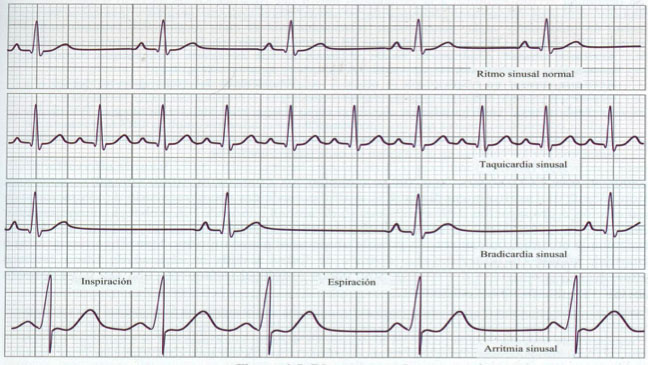
as arritmias abarcan un espectro muy amplio de anomalías del ritmo cardiaco, en el niño. En el niño habitualmente existe una arritmia respiratoria que es fisiológica, que se puede detectar durante la auscultación cardiaca, derivada de los cambios de volumen producidos durante la inspiración y espiración. Si este fenómeno es muy marcado se debe realizar un ECG para determinar si estamos ante una verdadera arritmia.
Ante una arritmia además del ECG debemos hacer una anamnesis adecuada. Hay que saber si existen antecedentes familiares de muerte súbita o arritmias tratadas, así como los síntomas que produce en el niño, si hay mareos frecuentes, dolor precordial, sincopes o palpitaciones. Los síntomas y signos de las arritmias son debidos a los efectos que ejercen sobre el gasto cardíaco, la cardiopatía de base y la edad del paciente.
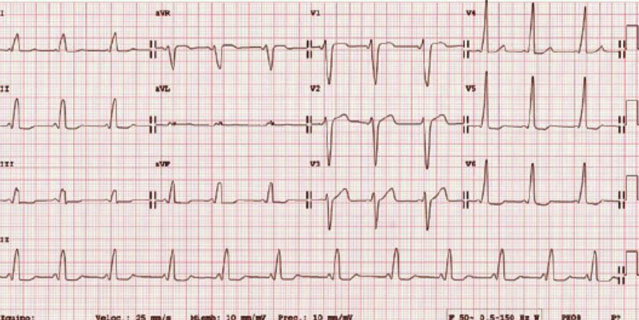
ECG de niño con SDM de WPW, se observa el intervalo PR corto y la onda delta que provoca ensanchamiento del QRS (en precordiales izquierdas; vía accesoria derecha).
Ante la sospecha de arritmia, el ECG de 12 derivaciones ofrece una información sobre el ritmo de base del paciente, la conducción, la repolarización, la evidencia de hipertrofia y otros signos. Las alteraciones en el ritmo de base pueden ser de mucha ayuda para la aproximación diagnóstica.
De gran importancia para el diagnóstico es obtener un ECG durante el episodio arrítmico. Cuando la arritmia es corta y esporádica es difícil conseguir el ECG, por lo que podrá ser necesario un Holter de ECG, que registra el ritmo durante 24 horas. En la actualidad se puede realizar un Holter de eventos (de más larga duración, de incluso 2-3 semanas) o Holter implantable para el diagnóstico y están apareciendo wearables para el diagnóstico (camisetas, relojes, dispositivos que se pueden añadir al Smartphone, etc.)
Las arritmias más frecuentes en los niños son las extrasístoles, que son latidos aislados diferentes al ritmo normal. Las extrasístolespueden ser auriculares o ventriculares, se debe descartar la probabilidad de que desencadenen taquicardias. Las extrasístoles ventriculares son las que pueden preocupar si son muy frecuentes, cuando son de morfología similar (monomórficas) y desaparecen con el ejercicio tienen criterio de benignidad, en esos casos hay que descartar patología cardiaca y deben ser revisados por un cardiólogo pediátrico.
Dentro de las arritmias destacamos:
TAQUICARDIAS
Ante un ECG con taquicardia, para un adecuado manejo debemos buscar el origen de ésta (auricular, nodo AV o ventricular). Las taquicardias auriculares se tienen que diferenciar de la taquicardia sinusal. El flutter o la fibrilación son taquicardias menos frecuentes en el niño que en el adulto.
Las taquicardias supraventriculares (TSPV) se producen por dos mecanismos que las diferencian:
- Reentrada nodal (Taquicardia Reciprocante AV Nodal), en niños mayores, por doble sistema de conducción dentro del nodo AV.
- Reentrada por vía anómala (Taquicardia Reciprocante AV Accesoria), principal mecanismo de los síndromes de pre-excitación (Wolf-Parkinson-White). Se trata de una vía accesoria localizada alrededor del anillo tricuspideo o mitral y que conecta directamente el musculo auricular y ventricular. Se caracteriza por observarse en el ECG un ensanchamiento inicial del QRS que se denomina onda delta y el intervalo PR corto por la preexcitación del ventrículo por la vía accesoria. Se diferencia entre taquicardia ortodrómica (la más frecuente, con QRS estrecho, sin onda delta y P retrógrada) y antidrómica (taquicardia QRS ancho por onda delta con P anterógrada).
- Otras causas de taquicardias son por automatismo anormal (menos del 10%) sobre todo en la taquicardia fetal, es producida por impulsos en tejidos que normalmente no tienen esa capacidad
Existe una forma especial de taquicardia por reentrada mediada por una vía accesoria con conducción decremental, es la llamada taquicardia reciprocante incesante de la unión AV –tipo Coumel-, se manifiesta como taquicardia lenta (120-180 lpm) persistente, incesante, con QRS estrecho y de muy difícil control farmacológico que requiere en la mayoría de ocasiones un tratamiento de ablación con radiofrecuencia.
Las Taquicardias paroxísticas supraventriculares originadas en la unión AV son las más frecuentes en adolescentes y adultos y la Taquicardia por reentrada por via accesoria en el niño pequeño. Se manifiestan a alta frecuencia (200-300 lpm), son de QRS estrecho. Responden a maniobras vagales y fármacos que enlentecen la conducción por el nodo AV. Clínicamente se toleran bien, pero cuando se prolongan en el tiempo pueden presentar signos de ICC (típico en el recién nacido y lactante pequeño). Se puede diagnosticar durante la gestación y se trata a la madre con fármacos que atraviesan la barrera placentaria.
Ante una taquicardia de QRS ancho podemos estar ante una taquicardia de origen ventricular (TV), son menos frecuentes que la TSPV. Ante una taquicardia ventricular con morfología de bloqueo de rama derecha es importante descartar la taquicardia polimórfica catecolaminérgica familiar y la displasia arritmogénica del VD. En estos casos debe tratarse al paciente con beta-bloqueantes o sotalol, o plantear ablación con radiofrecuencia e incluso el implante de un desfibrilador automático en casos con riesgo de muerte súbita. Existen casos de TV en pacientes con síndrome de QT largo, en los postoperados cardiacos con antecedentes de ventriculotomía y en miocarditis o miocardiopatías.
El tratamiento tiene como principal objetivo interrumpir la arritmia, enlentecer la respuesta ventricular y reestablecer un adecuado ritmo sinusal. En función de la situación clínica del paciente serán necesarias medidas de Reanimación Cardio-Pulmonar (RCP). Ante un paciente inestable, con gran compromiso hemodinámico, se recurrirá a la cardioversión eléctrica en primer lugar. En las TPSV se realizarán maniobras vagales (la más eficaz en el niño es mediante sofocación con hielo), seguido de la administración de Adenosina.
ADENOSINA
- Dosis de ataque: 0,1 mg/kg IV en embobada muy rápida.
- Si no hace efecto repetir el bolus en dosis crecientes hasta la dosis máxima.
En las TPSV se mantendrá tratamiento médico de mantenimiento (habitualmente betabloqueantes, flecainida y amiodarona de rescate) y en la actualidad, dado el bajo riesgo de la ablación se realiza este procedimiento en los pacientes que requieren tratamiento a largo plazo, con recaídas tras la supresión de éste o si son frecuentes a pesar del tratamiento. La ablación consiste en aplicar calor o frio (crioablación) sobre la zona de la vía anómala para cortar la vía de conducción que provoca la arritmia.
En las taquicardias ventriculares se debe tratar la enfermedad de base y en ocasiones pueden precisar un desfibrilador automático implantable.
Lectura recomendada:
BRADICARDIAS
Es la frecuencia baja respecto a la edad normal, hay que diferenciar la bradicardia anormal de la bradicardia sinusal, causada por determinados estados sistémicos (hipotirodismo, hipotermia o fármacos). La disfunción del nódulo sinusal es muy poco frecuente en el niño sano, se caracteriza por bradicardia sinusal, incompetencia cronotrópica al ejercicio y pausas sinusales. Se observa típicamente en el postoperatorio de cardiopatías congénitas corregidas.
El bloqueo aurículo-ventricular (BAV) es otra de las causas de bradicardia. Existen 3 tipos de BAV: el BAV de primer grado que no provoca bradicardia, consiste en un alargamiento del intervalo PR, el BAV de 2º grado que se divide en tipo I (alargamiento del intervalo PR de manera progresiva hasta que un onda P no conduce, es el llamado efecto Wenckebach) y el tipo II que de manera constante una onda P no es seguida por una contracción ventricular y, por último el BAV de tercer grado o completo, éste es raro en niños (1 de cada 20.000 nacidos vivos) pero es potencialmente letal. Típicamente se observa una disociación entre las frecuencias auricular y ventricular. Puede ser congénito (hijo de madre con lupus eritematoso sistémico o enfermedad autoinmune por anticuerpos anti-SS-A/Ro y anti-SS-B/La circulantes) o adquirido (complicación de cirugía cardíaca o ablación o secundario a infección como miocarditis viral o enfermedad de Lyme).
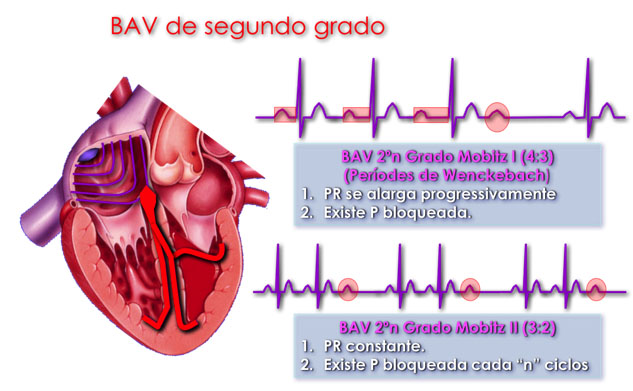
El manejo neonatal del BAV pasa por el ingreso del neonato para monitorización. Cuando la frecuencia ventricular es baja, el uso precoz de isoproterenol (0,1 mcg/kg/minuto) puede ser útil, pero el tratamiento definitivo es la colocación de un marcapasos.
Los marcapasos son el tratamiento de elección ante una bradicardia clínicamente sintomática. El objetivo principal del marcapasos es conseguir un ritmo cardíaco lo más fisiológico posible. En el niño pequeño se colocarán los cables epicárdicos (mediante toracotomía) y en los mayores se colocarán cables endovasculares (hay que tener en cuenta la necesidad de recambio de los catéteres conforme crece el niño).
El uso de marcapasos y DAI es cada vez más extendido en la población pediátrica, por lo que es importante que los pediatras de cabecera conozcan las precauciones que deben seguir estos pacientes, para cuidar de su marcapasos (MP). Su actividad se ve restringida sólo a los deportes de contacto. Para el resto de restricciones, dependerá de su función cardíaca, más que por el MP en sí.
Es aconsejable la profilaxis de la endocarditis bacteriana en los portadores de cables endovasculares. Se desaconseja la resonancia magnética, aunque cada vez más existen marcapasos reso-compatibles.